Jean Dessolin
Lauréat d'un AAP collaboratif 2021 nous présente son projet de recherche
Collaboration Dr. Valérie Desvergnes (ARNA)/ Dr. Jean Dessolin (CBMN)
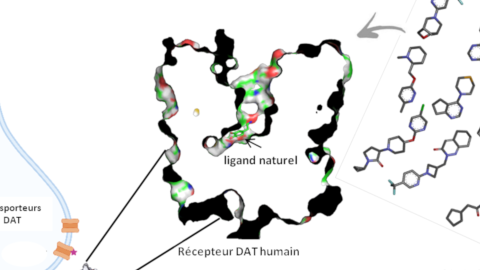
Dans le contexte d'un projet portant sur la recherche de composés actifs contre la maladie de Parkinson, la collaboration entre les deux équipes visait à chercher comment cibler les neurones dopaminergiques de la substance noire. L'approche a consisté à cribler une chimiothèque 3D afin d’identifier des ligands potentiels d'une protéine de la membrane des neurones : le transporteur DAT. En effet, les transporteurs DAT jouent un rôle essentiel dans la recapture de la dopamine dans le neurone présynaptique.

Cette méthode permet d’attribuer des coordonnées 3D à une protéine (ou un acide nucléique) à partir d’un modèle connu et assez proche. Les protéines DAT de drosophile et humaine partagent 52% d’identité de séquence (69% d’homologie), ce qui rend l’approximation tout à fait cohérente. Cette structure a été comparée à celle proposée par AlphaFold montrant peu de différences entre les modèles. Avec une structure tridimensionnelle du récepteur d’intérêt, il devient possible de rechercher des ligands de ce dernier. Le terme de criblage virtuel signifie que l’on a traité un grand nombre de molécules (des milliers) en tentant de les faire s’adapter à une cible, dans notre cas le modèle de DAT humain. Plusieurs méthodes existent, dans notre cas, seul le docking moléculaire qui simule le positionnement d’un ligand putatif dans la cible puis mesure l’énergie du complexe obtenu par le biais de logiciel(s), a été utilisé. Il est alors possible de comparer les énergies obtenues avec les différentes molécules et de les classer en fonction de leur affinité pour la protéine qui est appelée « le récepteur ». Cette technique demande donc aussi une banque de molécules, les « ligands » dans un format informatique reconnu par le logiciel de docking. Un grand nombre de chimiothèques est librement accessible sur internet, il reste ensuite à les préparer dans le format requis afin de les cribler et éventuellement de les filtrer en fonction des propriétés physicochimiques (Mw, hydrophobie, accepteurs/donneurs de liens H, etc…). Dans le cas présent, on s’est attaché à utiliser une chimiothèque commerciale afin d’avoir un accès simple et rapide aux molécules organiques sélectionnées par le calcul. Cette banque de composés étant déjà calibrée pour le passage de la barrière hémato-encéphalique, pré-requis à l’action sur le récepteur DAT, il n’a pas été nécessaire de la filtrer avant son utilisation. On aurait pu également utiliser n’importe quel ensemble de composés organiques en 3D et les filtrer en fonction de leurs caractéristiques souhaitées avant le criblage virtuel. Ainsi, la conformation et le positionnement de 9864 molécules ont été simulés dans le site actif du modèle produit. Après calcul des énergies de toutes ces solutions, un tri des composés a été effectué, limitant à 153 molécules la liste des composés qui seraient des ligands du récepteur ciblé. L’examen de cette liste permet de dégager un nombre restreint de motifs communs ("scaffolds") qui pourraient constituer autant de plateformes dans la conception de nouveaux ligands. La suite du projet consistera à intégrer par synthèse chimique certains de ces ligands potentiels sur des molécules actives de type nucléolipides afin d'évaluer leur capacité à cibler les neurones dopaminergiques.